
November 2, 2022
Finding the root cause of a manufacturing quality issue prevents unsafe or poor quality products from reaching consumers. Unfortunately, many investigations don’t get to the true root cause of a problem. As a result, mistakes continue, and poor product enters the market, increasing consumer risks and costs to remediate. Unfortunately, many FDA warning letters involve corrective and preventive action (CAPA) violations due poor root cause analysis.
This article covers common places manufacturers go wrong finding root causes of quality issues. In addition, it looks at how a quality management system (QMS) helps structure thorough investigations and prevent recurring issues.
Mistake #1: Not Digging Deep Enough
Finding the root cause of an issue is not always a one-step process. A top mistake manufacturers make is stopping investigations too soon. Without identifying the correct root cause, the problem is likely to persist.
For example, a 2022 FDA warning letter cites a pharma manufacturer for inadequately investigating out-of-specification (OOS) results. In that case, the manufacturer simply attributes the OOS result to analyst error. However, there was no review of their manufacturing process or an accompanying CAPA to inspect other batches.
While it’s easy to write off problems as human error, going deeper and asking why mistakes happen, you might uncover a deeper issue. For instance, there may not be a proper training program in place. Alternatively, there may not be a standard process for documenting OOS results. In these cases, human error is just a symptom of the true root cause.
Mistake #2: Using the Same Tool Every Time
Manufacturers have a variety of root cause analysis tools and models they can use for finding the root cause of an issue. Commonly used tools are often built into modern quality management systems, and charts are elements of a reporting dashboard.
Sample tools include:
- Pareto Chart
- 5 Whys
- Failure Mode and Effects Analysis (FMEA)
- Decision Trees / Fault Tree Analysis (FTA)
- Ishikawa Fishbone Diagram (IFD)
{kind=link}
The key is to avoid using the same tool for every issue. Instead, you should use the right root cause analysis tool for each situation.
For example, a decision tree is helpful when you have lots of historical data on previous problems, such as with mature processes. The 5 Whys method, conversely, is good for brainstorming potential root causes, or with simpler problems.
Mistake #3: Poor Documentation
Poor documentation of investigations is a common problem in many manufacturing organizations. FDA’s 2022 warning letters highlight the issue, particularly as it relates to investigations into OOS results.
FDA guidance recommends a full investigation whenever a manufacturer determines an OOS result is not caused by lab error. The investigation should follow a predefined procedure that includes a review of production and sampling processes. The agency also points out that it must be well-documented to include:
- A clear statement of why the investigation was launched
- Description of process elements that could have caused the problem
- The results of a documentation review and a likely or actual cause
- The results of a review into whether the problem occurred previously
- Description of corrective actions implemented
Mistake #4: Not Closing the Loop
A root cause analysis is not complete without expanding the investigation to determine if other lots or batches were affected. Not closing the loop in this way creates product safety and quality risks, as more bad product may be undetected. Only when you determine the full extent of the problem can you effectively prevent it from happening again.
As a best practice, manufacturers should create a closed-loop process between root cause analysis and corrective action that includes:
- Classifying the event as external or internal
- Managing risk to determine whether the issue needs a full review, as for high-risk items, versus a fast-track review for low-risk items
- Containment
- Corrective actions
- Verification and effectiveness checks
- Any adjustments needed
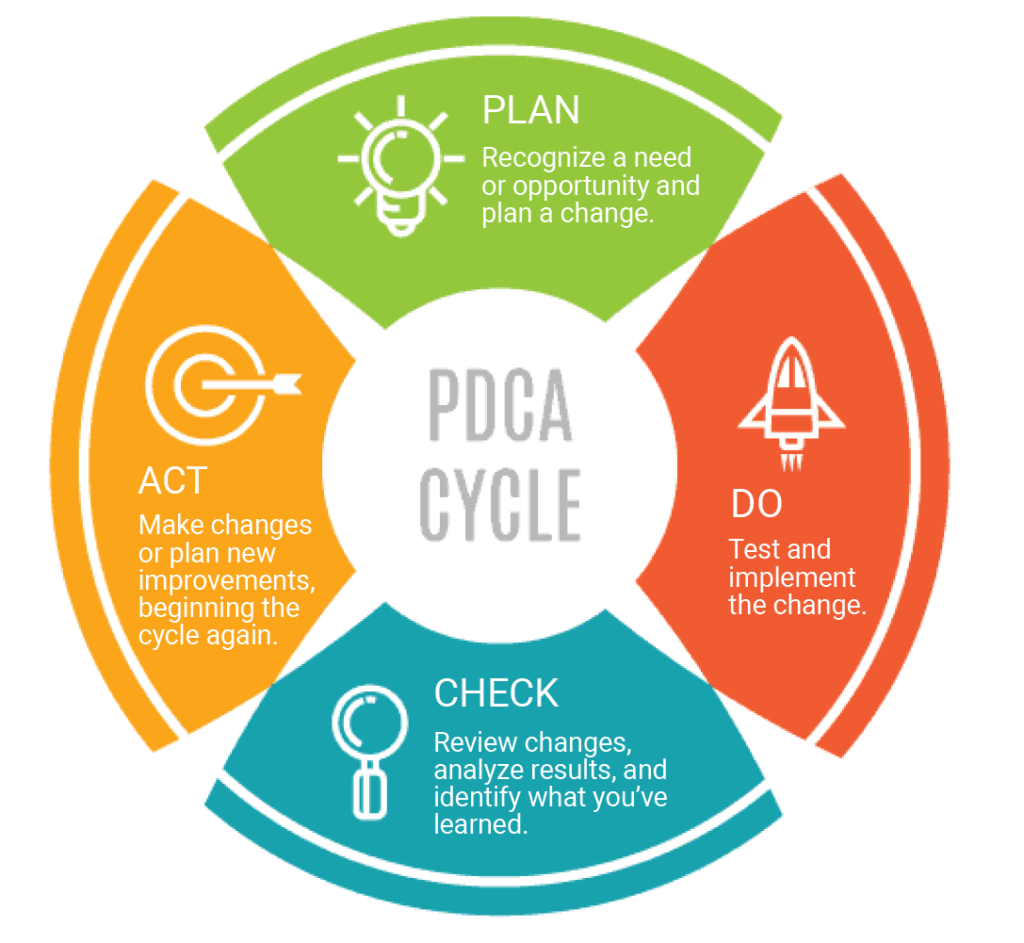 A closed-loop CAPA process closely follows the Plan-Do-Check-Act approach. PDCA is an iterative design and management method used for the control and continual improvement of processes and products.
A closed-loop CAPA process closely follows the Plan-Do-Check-Act approach. PDCA is an iterative design and management method used for the control and continual improvement of processes and products.
Mistake #5: Using Manual Quality Processes
Using paper or spreadsheets to conduct root cause analysis investigations creates gaps where issues can fall through the cracks. For instance, many people don’t use FMEAs correctly because they don’t know where they are and/or the FMEAs are out of date. To be effective, FMEAs need reviews for changes to production processes as well as recent corrective actions, complaints, or change control items.
The QMS provides a distinct advantage when it comes to root cause analysis investigations by helping companies to:
- Centralize process documents in a digital system to facilitate review, updates, and communication with regulators
- Link root cause analysis findings to corrective actions while creating a complete record of compliance activities
- Keep SOPs and investigation procedures updated and link any changes to employee training and change management processes
- Route, edit, and approve documents in a timely manner
- Ensure thorough investigation into whether other lots or batches are affected and need to be taken out of distribution
- Use data to identify trends useful in root cause analysis
Conclusion
Root cause analysis is fundamental to quality management, yet several challenges keep manufacturers from doing it right. Stopping at the symptom rather than digging deeper to the true root cause is a primary problem, as are poor documentation and follow-up.
Quality processes relying on paper forms and shared spreadsheets also increase the risk of recalls, regulatory penalties, and consumer impacts. An automated QMS accelerates resolution by tying together processes like root cause analysis, corrective action, document management, and change control. What results isn’t just a stronger root cause analysis process, but more reliable quality overall.
Related Reading: 5 Strategies to Improve Quality Incident Management and Root Cause Analysis
About the Author
Stephanie Ojeda is Director of Product Management for the Life Sciences industry at AssurX. Stephanie brings more than 15 years of leading quality assurance functions in a variety of industries, including pharmaceutical, biotech, medical device, food & beverage, and manufacturing.