
April 25, 2023
European Union (EU) Medical Device Regulation (MDR) and In-Vitro Device Regulation (IVDR) raises the bar for device safety. However, ongoing challenges have resulted in a new extension of the EU Medical Device Regulation transitional deadline.
European health commissioner Stella Kyriakides was first to propose the extension in December 2022. The draft amendment was adopted in March 2023 as Regulation (EU) 2023/607. The Council of the European Union now allows notified bodies (NBs) and manufacturers more time to certify medical devices. The intent of the extension is to mitigate the risk of device shortages that could result from a shortage of NBs.
EC members support the extension due to concerns over notified body capacity, fearing widespread shortages under the previous deadlines. Below we examine the changes, what’s driving them, and how manufacturers can meet EU MDR compliance challenges.
The Current State of EU Medical Device Regulation
The European Commission amended the regulations in March to postpone deadlines and avoid a shortage of life-saving products in the EU. It removes previous sell-off dates to prevent the unnecessary disposal of safe devices.
There are about 500,000 devices the new regulations impact which may not be compliant under the new rules. Furthermore, many devices have also been re-classified into a higher risk class under the new regulations. As a result, all devices must be resubmitted for compliance.
The complexity of new regulations, coupled with a very lengthy certification process and a scarcity of notified bodies (NB) will continue to create delays. As of April 2023, there are only 35 NBs to do the certifications, according to the European Association for Medical Notified Bodies. Today, the current backlog makes it unlikely for many devices to be certified on time.
MDR certification is proving to be time-consuming, 13-18 months on average, according to a report by MedTech Europe, the European trade association for the device industry. The group and many industry members have concerns that some legacy devices might be unavailable in the region because of certification delays.
Amendments to MDR and IVDR: Highlights
1. The Transition Period Focuses on Device Risk
The transition period was originally scheduled to end in May 2024. Now, a new sell-off provision allows medical devices on the market or still in the supply chain to remain available until May 2025.
The Medical Device Regulation extends the deadline for both higher medium- to low-risk devices including:
- December 31, 2027 for class III and class IIb implantable devices, with the exception of exempted devices such as sutures, staples, and certain dental devices
- December 31, 2028 for class IIa and class IIb devices not covered by the above, as well as class I devices in sterile condition or with a measuring function
- December 31, 2028 for devices with a declaration of conformity dated prior to March 26, 2021
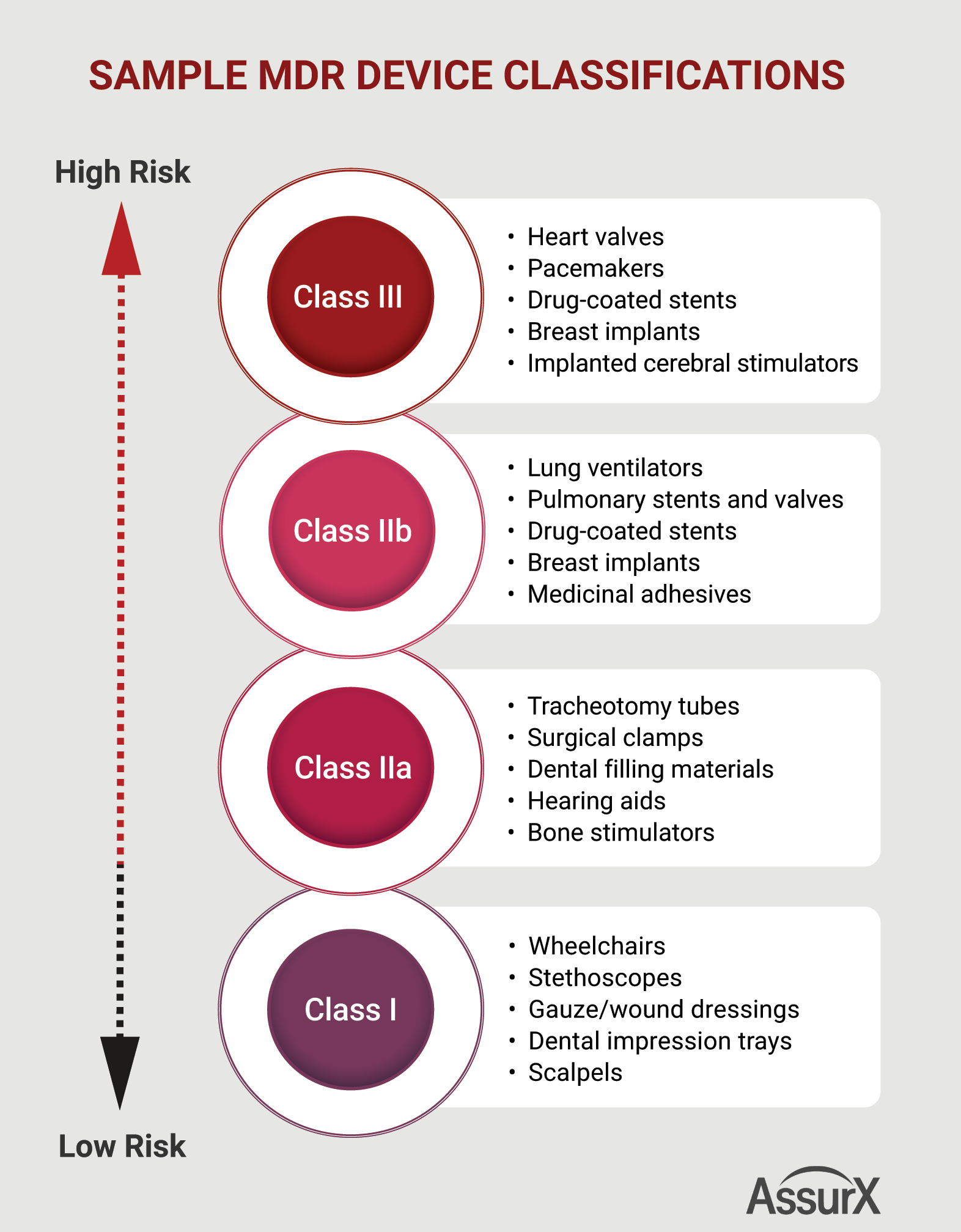
2. New Deadlines Allows More Time, but Time Shouldn’t be Wasted
The amendments to MDR and IVDR are meant to give NBs – not manufacturers – more time. The extension gives NBs more time to conduct conformity assessments. The council emphasizes that the extension aims to ensure a high level of public health protection, including patient safety, as well as averting any shortages of medical devices.
MDR and IVDR are already in effect and remain applicable. Therefore, the requirements for manufacturers remain the same. Device firms have been preparing for changes since the transition period began in 2017. Therefore, they should be ready now, and applications for certification should be submitted to NBs sooner than later.
For manufacturers, taking steps to ensure they have a QMS that complies with MDR before submitting is a critical priority. In fact, applying for a conformity assessment is considered an implicit confirmation that the manufacturer has an MDR-compliant QMS.
3. The Transition Only Applies to Legacy Devices
Under the amendments to MDR and IVDR, “legacy” refers to devices that were certified as compliant before May 2021. The application of the extended transition period is subject to several conditions, ensuring that only devices that are safe, and for which manufacturers have already taken steps to transition to MDR, will benefit from the extension.
New devices and legacy devices with a significant change in design or intended purpose are not covered by the extension, according to a notified body. Those devices should be compliant with the MDR when they first enter the EU market.
It’s important to note that there is a caveat for transition extensions for Class III custom-made implantable devices. Class III custom-made implantable devices (e.g., patient-specific bone implants) can be placed on the market without the relevant certificate until May 26, 2026. However, the manufacturer must lodge an application with an NB for conformity assessment by May 26, 2024. The manufacturer should sign a written agreement with the NB no later than Sept. 26, 2024.
Current Manufacturer Challenges Around EU MDR
A significant factor in the slow transition has been the steep learning curve around EU MDR compliance. Notably, technical documentation has been an application weakness, according to data from a survey of notified bodies.
Compared to previous regulations, the new MDR requires greater detail in a device’s technical documentation. Technical documentation includes extensive detail to demonstrate conformity with the regulation. Furthermore, documentation must contain both premarket and post-market conformity substantiation.
Premarket activities are documented in six chapters:
- Device description and specifications
- Information supplied by the device manufacturer
- Design and manufacturing information
- General safety and performance requirements (GSPR)
- Benefit-risk analysis and risk management
- Product verification and validation
Required documentation of post-market activities includes:
- Post-market surveillance (PMS) plan
- Post-market clinical follow-up (PMCF) plan
- Periodic safety update report (PSUR)
The new requirements around the PMS can be challenging for manufacturers. First, they demand a systematic approach to identify and manage risks based on quality data, including incidents, corrective actions, and complaints. Also, they require manufacturers to have processes in place to address data collection procedures, benefit-risk analysis indicators, threshold metrics, and quality process documentation.
The Importance of Risk Management from an EQMS Perspective
Risk management is key to compliance with the new regulations. Under MDR (Article 10), manufacturers are required to establish, document, implement, and maintain a risk management system. The higher the potential risk of a device, the more proof of its safety is going to be required. For example, device risks can stem from product variability, interaction with end users, device misuse, and device failure.
Here, it’s advisable that manufacturers incorporate risk management within the quality management system (QMS) to identify and mitigate risks throughout the product lifecycle. Furthermore, Medical Device Regulation emphasizes continual improvement of the QMS to ensure that compliance is proportionate to risk. Finally, in terms of risk management, the MDR requires sufficient financial coverage for potential liability that’s proportionate to the risk class.
Risk management requires more time, effort, and vigilance for device companies still using a manual or hybrid QMS. Switching to an automated enterprise QMS can make MDR and IVDR compliance easier, more efficient, and more effective. An EQMS provides a single, centralized platform connecting all quality processes that could impact risk management. It provides the transparency and the right tools needed to track and monitor quality issues for timely mitigation.
The Enterprise QMS for MDR Compliance
An automated QMS provides several advantages in the context of medical device regulation compliance.
Integrating solutions such as document management, complaints, corrective action, and reporting, creates a connected system that contains any type of documentation, as well as structured reporting and metrics for real-time tracking and trending. Furthermore, teams can incorporate risk management tools like failure mode and effects analysis (FMEA) to evaluate new and existing processes and systems.
In addition, a quality management system should provide manufacturers with the ability to submit Manufacturing Incident Reports (MIR) according to EU Vigilance guidelines and reporting time frames. Within the QMS, applicable MIR data is collected through the lifecycle of the medical device or drug delivery system. Centralizing data creates a controlled, systematic path for downstream activity such as post-market investigations and reporting as well.
Manufacturers can also link QMS data sources directly to regulatory requirements, which helps to:
- Streamline the process of creating controlled documentation
- Ensure consistent use of data and metrics for improved data quality and data integrity
- Provide a single source of truth when regulators or notified bodies have questions
- Create a complete, controlled compliance history with the ability to update technical documentation throughout the product lifecycle
- Ensure timeliness of MIR submissions
Conclusion
The extension of MDR amendments is intended to mitigate the risk of device shortages. However, the bandwidth and limited number of notified bodies will remain a critical factor. Manufacturers should be cognizant of the backlog and get ahead of submission deadlines.
Companies using an automated QMS to streamline MDR compliance and documentation are already one step ahead. Linking together many essential pieces of the process, the QMS provides a structured framework for compiling documentation and compliance activities. It also minimizes data quality and data integrity issues, while demonstrating to auditors a commitment to quality and risk management.
With these pieces in place, organizations can submit applications with confidence, both in their documentation and their processes overall.
About the Author
Stephanie Ojeda is Director of Product Management for the Life Sciences industry at AssurX. Stephanie brings more than 15 years of leading quality assurance functions in a variety of industries, including pharmaceutical, biotech, medical device, food & beverage, and manufacturing.