
January 16, 2018
Recent FDA warning letters indicate that many drug manufacturers do not have manufacturing in a state of cGMP control. In the first half of 2017, the FDA cited adulterated products and unsanitary conditions as the two most common violations in drug manufacturing. This article explains four ways an automated quality and compliance management system builds control into the production process with respect to adulteration and potential contamination.
1. Integrate eQMS to Improve Control
Two observations by the FDA note that (1) testing is often done only at the completion of a batch and (2) deviations found while in production were not corrected or documented properly. The common denominator here is a lack of control in the manufacturing process. Implementing an enterprise quality management system that integrates with other systems (EHS, supplier quality, batch management) will share information throughout the entire quality cycle. As a result, any nonconformance, regardless of origin, could initiate a new investigation through rules and workflows.
In the record below, the nonconformance in an automated QMS can be initiated by another system (a supplier, for example). The two systems communicate with each other and create one master record that becomes a single point of truth.
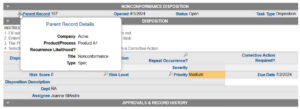
Sample automated nonconformance record.
In addition, consolidated reporting available in an eQMS enables trends and risk to be more readily identified. An eQMS with a robust measurements engine can evaluate data and trigger workflows for anything out of specification.
2. Implement an Integrated Document Management System
Adequate supporting documentation is required to evaluate control standards for all procedures. Without a central source of truth, documentation is unstructured, incomplete and time-consuming to compile.
A common observation by the FDA is lack of complete information for the manufacture of each batch prior to disposition. Consider what happens when a batch deviation is identified. Beyond your processes for ensuring that investigation, corrective actions and change management are executed efficiently, where is your supporting documentation?
A document management system is now a compliance best practice. Furthermore, having an electronic system designed for a QMS platform is the best solution. There are intuitive workflows already in place to automatically launch documents and revisions based on events happening across a quality system. Ultimately, one system contains and controls all records, revisions and change notifications which means greater visibility and transparency.
3. Build Risk Management into the System
The goal of any process should not just be to achieve desired results but to improve quality through monitoring systems and processes effectiveness. A continual process improvement methodology provides standards for identifying trends and measuring risk. As a result, data may yield areas of concern and opportunities to improve production quality and efficiency. Importing data into your QMS can provide unique insights such as trends and relationships in processes which may not be identified in other more traditional methods.
In the record below, a risk level has been assigned to a deviation. An automated QMS can prioritize records based on the risk score. Once dispositioned, signatures can be requested. Next, corrective action is added as a next step in the nonconformance investigation.
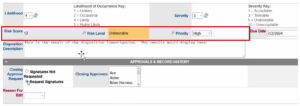
Automated risk measurement identifies and prioritizes critical events.
4. Conduct Routine Validation
A firm is required to prove it is operating within a state of control. Therefore, it must verify that cGMPs are in conformance. This includes conducting process validation to ensure that the data submitted is compliant and useful.
In this instance, it’s clear that a discrepancy exists. An automated system would begin an investigation based on a lot of rejection. By the end of the investigation, the firm will have statistical findings and documentation of the nonconformance. If necessary, a CAPA will automate correction of the nonconformance and re-validation of the process. Plus, an automated system can trigger any type of impurity study at any time during production to validate the reliability of the process. The result is evidentiary proof that the issue is resolved, and the new process is repeatable and meets all cGMPs.
Conclusion
Compliance means a firm has established suitable, adequate, and appropriate controls to yield pure and quality product(s). Leveraging a robust QMS platform enables firms to gain control of all aspects of the production process to detect and remedy adulteration. It uses systematic processes to provide evidence that controls are documented, monitored, measured and reviewed. Integration with multiple systems further extends workflow capabilities and builds a reliable, centralized source that provides a birds-eye view into the state of compliance. The end result is more control, unified management and higher output of a consistent, quality product.

Request an online AssurX QMS demonstration