January 13, 2014

Dennis Payton, Vice President of Product Marketing and Development, Expandable Software, Inc.
Some of the shortest descriptions in the FDA CFR 21 Part 820 Quality System regulation are found in Sec. 820.30 and Sec. 820.40 totaling about a page of written language around Design Controls and Document Controls. However short, these two sections can be the most complex aspects of Medical Device controls when actually complying with the regulation. Fortunately, the FDA does give a bit more background to help a new medical device company understand these two key elements (see Medical Device Quality Systems Manual, A Small Entity Compliance Guide) but again with the detailed complexities, even those few pages of guidance (covered in section 9 Document and Change Control) fall short of coverage needed to understand the impact a company’s Medical Device Quality System. The good news is that there are some very good tools that can help mitigate these complexities and streamline controls. The bad news is that it still takes a very strong detailed and sustained effort to ensure these complex controls are in place for continued success and compliance with the regulation.
Therefore with a wide variety of Medical Device suppliers there comes a wide variety of processes, procedures, and controls that are developed specifically for a business and to the Medical Device(s) being produced. It is important to understand how the FDA tries to normalize a specific business to the regulation when auditing that business for Design Control compliance. Having a bit of understanding of their view will help make for a much smoother comparison, analogy, and a much cleaner and successful audit of a company’s design processes.
A simplistic model can be derived from the 820.30 regulation that the FDA may use to assure design coverage and compliance of device design and/or manufacture to the regulation. The design and development model can be graphically depicted and loosely linked to the regulation as follows:
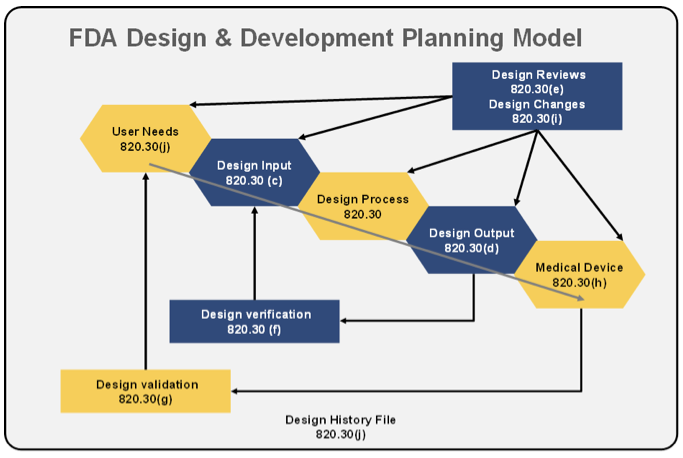
Diagraming out the design flow is helpful in seeing a more detailed picture of the flow and validation and verification of a product against its intended use model and specifically important to the FDA that each and every stage of the process is well-reviewed and documented.
Again, like the FDA regulation on Design Controls, this is a very short summary of complex processes, document definitions, controls, and general management & approvals that there have been volumes of books written. The objective should be to have a very good understanding of how the FDA or other regulatory entity views the medical device controls such that a business can demonstrate how their particular controls map into the regulatory model. A logical analogy of a business’ design and development model should be able to map to the regulatory normalized baseline model(s), in doing so, will result in smoother audits with a higher degree of success and hopefully (something the regulatory folks don’t really care about but as a business we all do) a lower expense/time in managing through the audit process.
However, a fuller descriptive paper outlines some key points in the development of a Medtech-specific design control with a product development process and how to maximize the use of enterprise-level business tools that accelerate the process, streamline audits and make for much smoother compliance. The brief outline here is a key element to more streamlined and smoother compliance with regulation keeping in mind not just the business drivers but also the FDA’s “normalized view” of design controls.
About the Author
Dennis Payton is Vice President of Product Marketing and Development with Expandable Software Inc. He has 24 years of engineering, product management, and executive management experience. He holds a BS in electrical engineering from California Polytechnic State University, San Louis Obispo, and post studies at Stanford University, University of California, Santa Cruz, and UC Berkley Haas School of Business.
Copyright UBM Canon. Used by permission.