
July 13, 2021
Corrective and preventive action (CAPA) violations are the most frequently cited non-compliance in FDA 483 inspection observations. In 2020, nearly 200 violations were cited among medical device manufacturers alone.
Unfortunately, those statistics indicate that not enough attention is given to CAPA management. Organizations leave themselves open for regulatory action, quality failures, and patient safety issues. Recently, onsite FDA inspections have seen a decline during the Covid-19 pandemic. However, device and diagnostics manufacturers can safely assume the agency will re-focus the lens on CAPA in both virtual and onsite audits.
This article looks at how to simplify CAPA management with a cloud-based quality management system (QMS). Implementing the right solutions and methodology enables alignment with FDA expectations while also protecting patients and the business.
Create a Closed Loop, Risk-Based CAPA Management Process
For medical device manufacturers, the most frequently cited compliance issue related to CAPA is inadequately documenting procedures. This issue resulted in 165 citations across 422 warning letters in 2020. Similarly, the next most frequent citation in this category is lack of adequate CAPA documentation, representing an additional 32 citations.
In many cases, these types of citations result from not completing a CAPA ̶ or not completing it right. To address this issue, manufacturers should be using an automated QMS that creates and documents a closed-loop digital CAPA management process.
Automating CAPA provides a controlled process that enables:
- Logging of issues in the CAPA system with notifications automatically sent to the appropriate parties based on risk and priorities
- Documenting any containment actions or on-the-spot corrections
- Initiating root cause analyses to get to the true source of the issue
- Creating an action plan and automatically notifying responsible individuals of their action items
- Verifying that the CAPA has an approved effectiveness check before closing it out
- Using business intelligence (BI) tracking tools like charts and graphs to detect trends and verify effectiveness of the corrective action
- Following up on the issue in the future by linking it to QMS functions such as employee training, audit management, change control, risk management, and new product development.
Transitioning from paper processes to a cloud based QMS centralizes communications, action items, and compliance history. Standardized forms, business rules and workflows ensure consistent processing that eliminates lost information while minimizing delays.
In addition, a compliance risk analysis should be performed. The results of the risk analysis should guide the CAPA timeline based on assigned risk. Furthermore, being able to provide end-to-end documentation of this closed-loop process is essential to audit readiness, and developing real world knowledge management.
Avoid Making Your CAPA System a Dumping Ground
Another common mistake is allowing CAPAs to accumulate, creating a huge backlog of issues of widely varying degrees of risk. According to the FDA’s Case for Quality CAPA working group, 70% of corrective actions observed were incorrectly classified and could have been managed with more simplicity.
Without a consistent process to identify CAPAs, assign by risk, and track progress, high-risk issues can get lost in a sea of CAPAs. This allows CAPAs to snowball into costly quality issues, missed improvement opportunities, and can result in regulatory audit findings.
Sometimes, companies hire a dedicated team just to help process their CAPA backlogs, some of which have been lingering for years. However, this approach requires a huge investment of time and energy that often creates business disruption and introduces more risk. Unfortunately, this practice is sometimes the only way to clear the backlog.
The first step in fixing this problem is to qualify and rank CAPAs based on risk assessment. Not every issue is a CAPA, and the corrective action process shouldn’t be a catchall term. Risk management requires clearly documented procedures for CAPAs, and that’s what the FDA looks for.
Prioritize CAPA Management by Risk
Simplifying CAPA management requires incorporating risk into each CAPA request. Risk and corrective action go hand in hand, and you can’t effectively manage one without the other.
Most importantly, your company needs to establish criteria for acceptable vs. unacceptable risk based on an assessment and risk calculations applied to historical data. Risk is a measure of severity and likelihood of occurrence, with a risk matrix highlighting these essential relationships. See Figure 1.
Subsequently, each CAPA request can then be assessed and identified according to its source and inherent risk to the product, the patient, and the business.
When you enter a CAPA in the QMS, you can use risk management tools to help consistently identify and filter items that require critical fast-tracking. Investigate high-risk issues first and establish preventive actions for preventing recurrence.
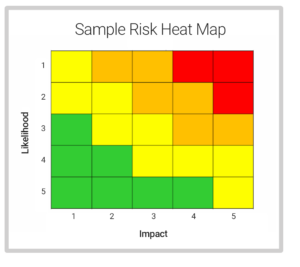
Figure 1: A sample risk heat map (also referred to as risk matrix). Likelihood and potential impact can determine risk for CAPA management.
Corrective Action vs. Preventive Action: What’s the Difference?
Corrective action focuses on fixing a single instance of a problem. Preventive action, however, goes a step further, implementing measures to stop the problem from happening in all processes, lines and plants.
Steps in a corrective action include:
- Shutting the machine or process down to isolate and contain the problem material
- Make corrections and approvals to restore the machine or process to service
- Quarantining the affected product for further analysis and/or disposition
- Notifying process owners and logging the event in the quality system
- Root cause analysis to determine the source of the problem
- Taking steps to prevent recurrence at the specific work area
- Conducting an effectiveness check to verify the correction and corrective action are working
Subsequently, preventive action is the steps you take to eliminate or minimize risk of potential occurrence, even if an issue hasn’t happened yet. Prevention isn’t always possible, but what’s important is communicating and applying the lessons learned across the organization. Getting people to start proactively thinking about prevention is the very definition of risk-based thinking. It’s getting people to ask, “Could that same issue happen in this process?”
Core QMS Solutions to Link with CAPA Management
Corrective action is a huge pain point for medical device manufacturers, especially those that receive FDA audit findings, 483 inspection observations or warning letters. However, deploying corrective action as the first process in a QMS implementation isn’t necessarily the best idea.
Instead, document control and employee training are more often a logical starting point for quality management software. Documentation and training are the basis of process standardization. Without both, proper documentation and employee training cannot align effectively with process documentation changes. For a CAPA process to be effective, these two processes should be in place first to ensure consistent performance.
Once document control, employee training and CAPA are in place, companies should turn their attention to other related processes such as:
- KPIs, critical-to-quality metrics and BI reporting
- Complaint handling
- Change control
- Supplier quality
- Risk management
Conclusion
CAPA processes (of lack thereof) have long been a cause of audit findings and quality problems, often because it is under-emphasized. From a practical perspective, you need to make people aware of it at every level—not just the quality department.
In addition, CAPA management is still in practice by many as a task to maintain compliance. However, when integrated with an automated QMS, CAPA forms a critical hub for investigating and closing the loop on problems with documentation and supporting data capable of withstanding audit scrutiny.
When deployed efficiently, a CAPA system is much more than a compliance requirement—it is a competitive business strategy. If can facilitate design changes, reduce product recalls, reduce rework and create other efficiencies that add up to quality cost savings, better quality and safer products.
