
June 30, 2022
The European Union (EU) Medical Device Regulation (MDR) officially went into effect in May 2021, while the In-Vitro Device Regulation (IVDR) took effect in May 2022.
The new legislation established the creation of EUDAMED, an online repository of information intended to streamline access to data for regulators, medical professionals and the public. Pandemic-driven delays, however, have extended timelines for MDR implementation, creating many questions about when companies must meet EUDAMED requirements.
Who must register as an actor? When must they register devices in the database? Most importantly, what they should be focusing on how to ensure compliance?
In this EUDAMED update, we examine elements, deadlines, and action items that device manufacturers should have on their radar.
What Is EUDAMED?
The European Databank on Medical Devices (EUDAMED) is part of the EU’s new Medical Device Regulation (MDR). Once fully implemented, it will provide data across the product life cycle for medical devices sold in the EU. Goals of EUDAMED are to increase transparency around medical device performance for the public and healthcare professionals and improve coordination between EU member states. It provides competent authorities with rapid access to relevant regulatory information. It also implements a uniform approach to Medical Device Directives.
Based on product classifications and applicable provisions MDR/IVDR, EUDAMED will contain the following information:
- The registration of manufacturers, Authorized Representative and devices
- Declaration of conformity
- Vigilance system
- Traceability process
- Labeling and instructions
- Justification of classifications (Annex IX)
- A copy of ISO certificate and/or Proof of QMS
- Information on Clinical Investigations
- Additional documentation as required based on device
Current Status of EUDAMED Modules
EUDAMED is comprised of six modules:
- Actor registration
- Unique device identification (UDI) and device registration
- Notified bodies and certificates
- Vigilance
- Market surveillance
- Clinical investigations and performance studies
Currently, only modules on actor registration, UDI and device registration, and notified bodies and certificates are available for voluntary use. The remaining three will go live once the EC declares all modules fully functional.
EUDAMED Update on Compliance Dates
EUDAMED use won’t be mandatory until all modules are operational and the European Commission has published a notice in its official journal. According to the proposed timeline and transition periods, mandatory compliance dates with each module will be:
- Q4 2023: Actors, Vigilance, Clinical Investigation and Performance Studies, and Market Surveillance
- Q2 2025: UDI and Device Registration and Notified Bodies and Certificates
Actor Registration
The Actor registration module has been live for voluntary use since December 2020. Once the EUDAMED system is fully functional, all economic operators must register as actors in the database. Economic operators include:
- Manufacturers in the EU
- Non-EU manufacturers
- Authorized representatives of organizations that are located outside the EU
- System/procedure pack producers
- Importers
The process involves submitting a registration request to receive a unique actor identifier/single registration number (Actor ID/SRN). If an organization has multiple roles, for example as both a manufacturer and importer, it will need a separate Actor ID/SRN for each. This will require separate registration requests.
UDI And Device Registration
Once EUDAMED is fully up and running, manufacturers must register all devices placed on the EU market with a UDI. The goal is to improve traceability and response to product safety issues by making it easier to locate devices within the supply chain.
EUDAMED update for UDI submissions:
- As of October 2021, manufacturers can voluntarily submit UDI/device information to EUDAMED.
- Since May 26, 2021, all Class III medical devices and implantable devices sold in the EU are required to have a UDI.
- By May 26, 2023, all Class IIa/IIb products must comply with UDI.
- By May 26, 2025, all Class I products must comply with UDI.
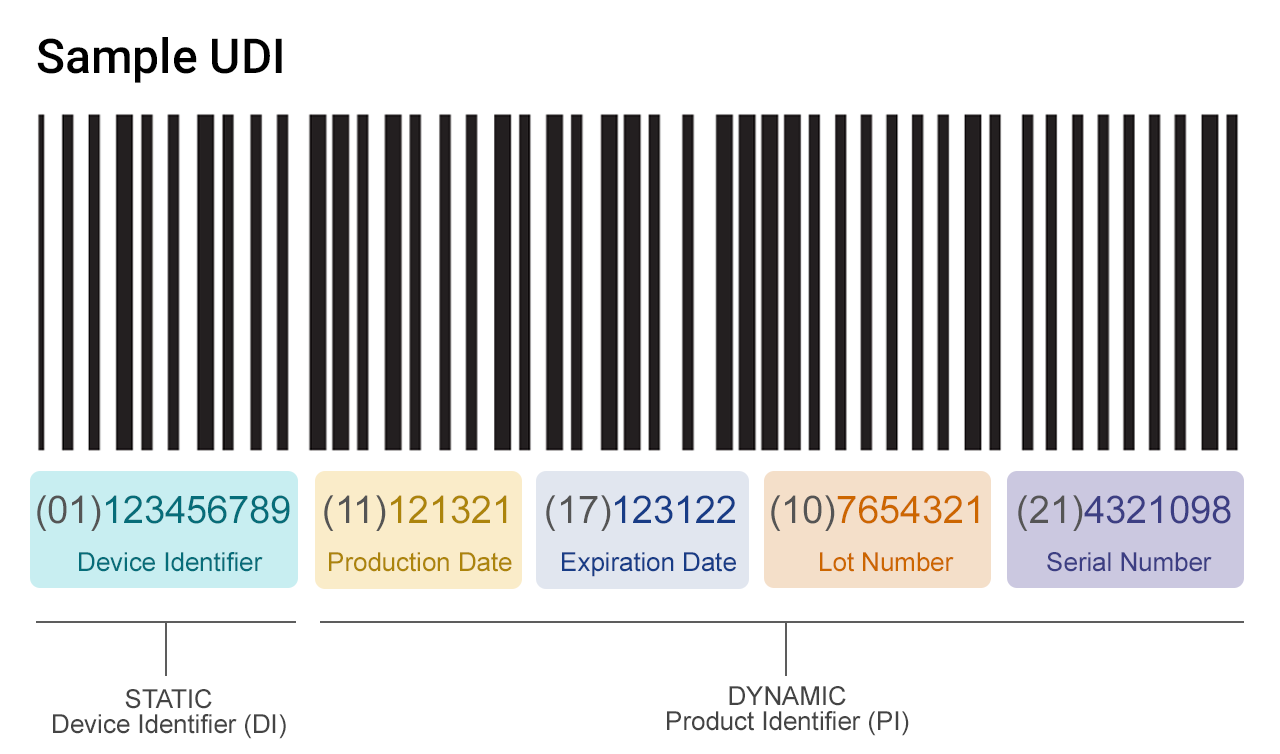
Manufacturers are advised to track product data including UDI within an electronic quality management system (QMS). Controlling this data at the product level is critical to compliance both with EU and FDA requirements, as well as any countries requiring UDI in the future.
Notified Bodies and Certificates
Under EU MDR, notified bodies (NBs) must register information on certificates in EUDAMED for public access. Since October 2021, NBs can voluntarily register certificates and Summaries of Safety and Clinical Performance (SSCP) so long as the parties the certificates reference are also registered. This includes:
- The device manufacturer
- The authorized representative
- The system procedure pack producer
- The device’s UDI
One reason for the long compliance timeline on this module has been the limited number of NBs available to certify devices. This is in part due to the change in relationship under the new device regulations. Previously, the relationship between the manufacturer and NB was more of a partnership, where the NB would help facilitate the process. Now NBs are expected to play more of a gatekeeper role, a shift that has required lengthy retraining of NBs.
Manufacturers should be aware of potential delays as NBs continue to work through their backlogs. Key action items are to understand where devices fit under the new classification, prioritizing devices for certification, and getting in queue with your NB.
The Biggest Hurdle: Market Surveillance
While manufacturers should already have processes for clinical investigations and vigilance, EU MDR’s post-market surveillance requirements are completely new. Unlike the previous MDD, MDR compliance requires going beyond complaint management to actually conduct post-market surveillance studies.
The reason is that complaints represent just a small slice of real-world data. Only a small proportion of patients and medical professionals with problem devices register complaints, providing insufficient data on product performance. Now manufacturers must collect more comprehensive data in a post-market surveillance (PMS) plan to better identify and manage risks of devices on the market.
Breaking Down the PMS Plan
The PMS plan is intended as a series of feedback loops used to continuously improve device safety and performance based on real-world data. The PMS plan must define the data that will be collected on each device, which may include but isn’t limited to:
- Incidents
- Corrective actions
- Trend reporting data
- Complaints
- Feedback
- Technical literature or databases
The plan must also cover:
- The process for collecting the above data
- Indicators and threshold values used in benefit-risk analysis
- Methods and processes for complaint investigation, communication, incident management, and corrective action
- Traceability tools and processes
It’s important to note here that the PMS plan can be complex, involving longer response times and multiple stakeholders. Add that up across multiple devices, and it’s clear companies need to be laying the groundwork now.
Conclusion
EUDAMED is slowly coming online after pandemic-related delays and an ongoing bottleneck in the number of available NBs. To be ready for compliance dates, manufacturers need to be thinking now about actor registration and UDI requirements, as well as how they will collect post-market surveillance data.
An automated QMS built around medical device manufacturing best practices can help:
- Centralize data for easy access and communication with regulators and NBs
- Track compliance requirements and associated documentation to minimize gaps and expedite MDR certification
- Compile and analyze data on complaints and post-market surveillance to proactively address device risks
- Prepare for future digital transfers to EUDAMED once it is fully approved to improve data quality, accuracy and speed of changes
On a larger scale, regulators globally are pushing the industry to better leverage technology to reduce patient risks. QMS automation is a key first step, ensuring quality data is systematically collected, managed and analyzed to continuously improve device performance, from development to post-launch.
About the Author
Sal Lucido is Co-Founder and Executive Vice President AssurX. Sal is an unequivocal product evangelist and an expert in the area of quality process automation. He holds a broad array of responsibilities, ranging from overseeing strategic plans and operational improvements to managing tactical alliances.