October 15, 2014
After a flurry of activity, it’s been relatively quiet of late on the FDA warning letter front. But three device makers did get some bad news in recent weeks.
FDA’s Philadelphia office hit Pittsburgh-based Zoll Manufacturing Corporation, a maker of Class III medical device Life Vest with a number of observations, including failure to document a corrective and preventative action program (CAPA), review, evaluate, and investigate complaints, adequately establish procedures for design validation, and develop, maintain, and implement electronic Medical Device Reports (eMDRs) to the agency.
The eMDR Final Rule requiring manufacturers and importers to submit eMDRs was published February 13, 2014. The requirements of this final rule will take effect August 14, 2015.
Zoll’s senior management was also singled out in the September 23 warning letter for being unable to provide records of adequate attendance at management review meetings in 2013 and this year.
Our west, FDA hit Mission Viejo CA-based Alpha Medical Instruments for CAPA and other violations. Alpha manufactures angiographic balloon catheters.
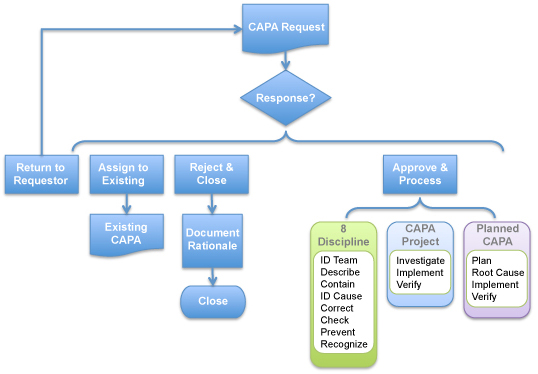 Digging deeper into Alpha’s alleged CAPA problems, FDA’s October 7 letter noted that the firm closed out an investigation without being able to produce documents proving it had conducted an effectiveness check. Additional product problems emerged after the CAPA was closed out. Alpha’s subsequent response was found wanting by the agency.
Digging deeper into Alpha’s alleged CAPA problems, FDA’s October 7 letter noted that the firm closed out an investigation without being able to produce documents proving it had conducted an effectiveness check. Additional product problems emerged after the CAPA was closed out. Alpha’s subsequent response was found wanting by the agency.
Alpha was also dinged for failure to establish procedures for reviewing, receiving, and evaluating compliance by a formerly designated unit, among other issues.
It’s worth noting that, while industry sometimes complains that FDA inspectors are inconsistent in terms of focus and expectations, it’s pretty hard to say FDA isn’t sticking with its emphasis on CAPA issues.
Finally, FDA identified problems of a different sort at Powers Device Technologies Inc., a Del Ray FL-based manufacturer of the Pacifier Activated Lullaby. The firm was charged in a September 25 warning letter with making unapproved product modifications and marketing those supposed new benefits without agency review. It’s been marketing the product as FDA-approved when, in fact, the agency said it has not approved the product as now constituted. The company must respond later this month in writing to the FDA to address these and other observations.