
May 30, 2019
FDA regulation 21 part 822 lays out the process, the rules, and the products that require postmarket surveillance. The process is an active, systematic, scientifically valid method of interpreting data or other information about marketed devices. More importantly, postmarket surveillance is an effective way to prevent and reduce problems that may impact public health. Despite regulations and guidance, inefficiencies and breakdowns are demonstrated in FDA warning letters and recalls. This article provides recommendations that can increase the success of a postmarket complaint handling process and provide deeper insights into quality throughout the entire product lifecycle.
Develop a Top-Down Culture of Quality
A quality culture can determine the effectiveness of a complaints management process. Product quality is driven by many moving parts outside of QA and RA departments. While these teams are accountable for quality processes and procedures, it would be short-sighted to think that a segment of an organization can be unilaterally successful at ensuring customer satisfaction and product quality. A culture of quality is integral in ensuring the implementation of quality throughout the company.
Broadening the organizational lens of compliance management is a critical task that should start with senior management. This group sets the tone for the entire organization. Managers can support departments by increasing communication of the importance of the compliance management process. Furthermore, they should reinforce the idea that compliance management is a continuous process and ensure that employees feel empowered and have the tools and controls they need to do their part in compliance management.
Implement Design Controls that Meet Risk-Based Standards
Design controls are an essential component of a quality system. They set forth the standards and expectations to control premarket and post-market design processes while ensuring that product specifications and intended uses are met. In addition, design controls should contain criteria for evaluating risk and quality in the development stage. However, design controls continue to remain a common subject of FDA Warning letters.
In 2019, 40% of FDA warning letters warning to device manufacturers cited deficiencies and failures in design control documentation, risk assessments and/or validation. Lack of insight and substantiation of product design and testing leaves a giant gap when a complaint requires root cause analysis. It is essential to document design, validation, change, and risk assessments.
As a litmus test, look at real examples of validation test records within your own company. Find your own failures and check to see how they are followed up on. An effective validation process seeks to “close the loop” on validation mistakes.
Institute a risk-based approach to change—assess the risks associated with each change and take action accordingly. Every change made to a product introduces risks that must be minimized or eliminated. As companies develop and document the change management process, they must also go back and audit the process, as well as ensure those audits are recorded for evidence of integrity.
Develop Thorough Procedures for Deviations and Nonconformance
One of the most effective ways to improve quality is to tap into technology to ensure the manufacturing process is effective. At the most basic, technology can be used to set up due date reminders to ensure deliverables are on time. Further, manufacturing teams can work with quality to set up enforcing algorithms or rules can ensure important information is collected and acted upon.
For example, say there is a decision to accept a nonconforming product as is. The quality team can make it a mandatory rule that whenever a “use-as-is” decision is made, management is alerted or that some rationale is required. Such steps ensure that decisions are documented and reviewable. Further, it ensures that questionable rationale such as “we can’t miss our deadline” or “we need to save money,” is avoided.
Monitor the effectiveness of the systems by setting up key performance indicators. These indicators set quality goals and monitor progress to those goals, and downstream, provide irrefutable data when a CAPAs are required for adverse events.
Integrate Supplier Management and Purchasing Controls
Section 21C of CFR 820.50 defines the quality system requirements for purchasing controls. It requires that each manufacturer establish and maintain procedures to ensure that products and services are specified to conform to the requirements. Organizations have a duty to ensure suppliers are qualified and certified to meet the manufacturing or service needs before purchasing a product from them.
However, FDA warning letters document significant violations related to management and monitoring of suppliers and supplied product. The number of such violations indicates an industry-wide lack of proper evaluation of supplier contracts and suppliers.
Further, regular audits of suppliers can help ensure they maintain compliance. An effective way to do this is to set up a schedule and adjust it for frequency based on the audit results—that is, suppliers who need more oversight should have audits more frequently.
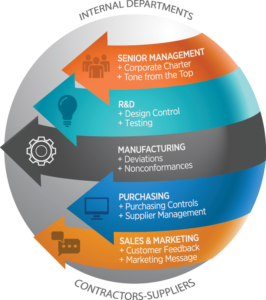
A comprehensive complaint management process facilitates the gathering, evaluating, investigating and remediating complaints by connecting insights from all areas of the organization that can impact issue investigation, remediation, and closure.
Don’t Underestimate the Role of Sales and Marketing
Sales departments play a role as a customer connection, crafting the marketing message, and ensuring that feedback is received. Perhaps most critical to complaint management, sales teams are on the front line to help facilitate postmarket surveillance.
Sales and marketing are both proactively passively receiving product feedback daily that includes complaints. An effective postmarket surveillance system allows this group to share the information received from customers for further evaluation it to determine if valid complaints exist and require an issue to be opened.
The same applies to field service personnel, who are often conduits for important product information. There is often a wealth of information in data collection from the field. Use this data as part of overall postmarket oversight to reveal trends that might be going unnoticed. If a problem is happening consistently and being overlooked, it is probably not being documented properly.
It is important to ensure processes are documented. But it is also key to train, audit and evaluate the overall effectiveness of the process. Ultimately a firm should be able to show and prove that there’s a continuous improvement system in place.
Conclusion
Quality and compliance should be a guiding principle of enterprise operations. The effectiveness of a medical device complaint handling process is determined by the processes in place to guide quality, which includes a scope of engagement that includes multiple business segments. An important way to create a good quality system is to take a risk-based approach to compliance management throughout the product lifecycle, from design and development through manufacturing and release. By expanding the scope of the processes, involving all stakeholders, and taking advantage of technology, companies can get ahead of problems. Keep in mind, these steps require a watchful eye to ensure that a closed-loop continuous process is in place and that it is effective.
