
January 19, 2021
With the May 26, 2021 implementation deadline looming for the new EU Medical Device Regulation (MDR), medical device companies are under pressure to ensure products are compliant with the new law.
There’s only one problem. The time it’s taken to train notified bodies (NBs) on the new MDR and IVDR requirements. This has created a bottleneck in available resources that could delay certification for companies.
The same problem extends to IVDR compliance. Despite the In-Vitro Diagnostic Regulation (IVDR) delay to 2022, many companies are going through the certification process for the first time with previously unclassified devices that are now regulated.
For medical device and diagnostic companies, the implications could mean big changes in which devices are marketed in the EU. Let’s look at what companies need to know, how to cope with the challenges, and how companies of any size can improve audit readiness and efficiency.
Reasons Behind the MDR and IVDR Certification Slowdown
EU MDR represents a fundamental shift in how medical device companies and Notified Bodies (NBs) work together. In the past, the NB served as a partner in helping companies get devices approved. However, the new role of the NB under MDR focuses more on guidance and even enforcement via unannounced audits.
In essence, the relationship that existed between manufacturers and NBs is gone and is now based on an entirely different set of regulatory rules, requiring their recertification.
Retraining NBs on the changes has reduced the overall number of organizations available to help device companies needing to register products under the new law. To date, the European Commission has certified 18 NBs to MDR, down from 21 certified under the previous MDD. While the number of NBs has grown in recent months—up from 11 in September 2020—it’s taken a while to get them online since MDR went into force in 2017.
This long timeline for getting NBs on board and the resulting shortage of resources is the main reason why EU MDR implementation was delayed a year from its original compliance deadline of May 2020.
The Coronavirus crisis and the ensuing loss of regulatory expertise due to staffing cuts have also contributed to the delay. The result is that some companies have been scrambling to replace subject matter experts to prepare for May 2021, inevitably leading to a need for external consultants.
Understanding Which Devices Are Covered Under MDR and IVDR
Adapting to the bottleneck in resources first requires understanding what you need from your NB. To that end, companies will need to start by determining which of their devices need to undergo the registration process based on new classifications and deadlines.
Some devices that weren’t previously considered medical devices, such as contact lenses, are now regulated under MDR. In other situations, the classification of the device may have changed, with almost all devices needing more investigations.
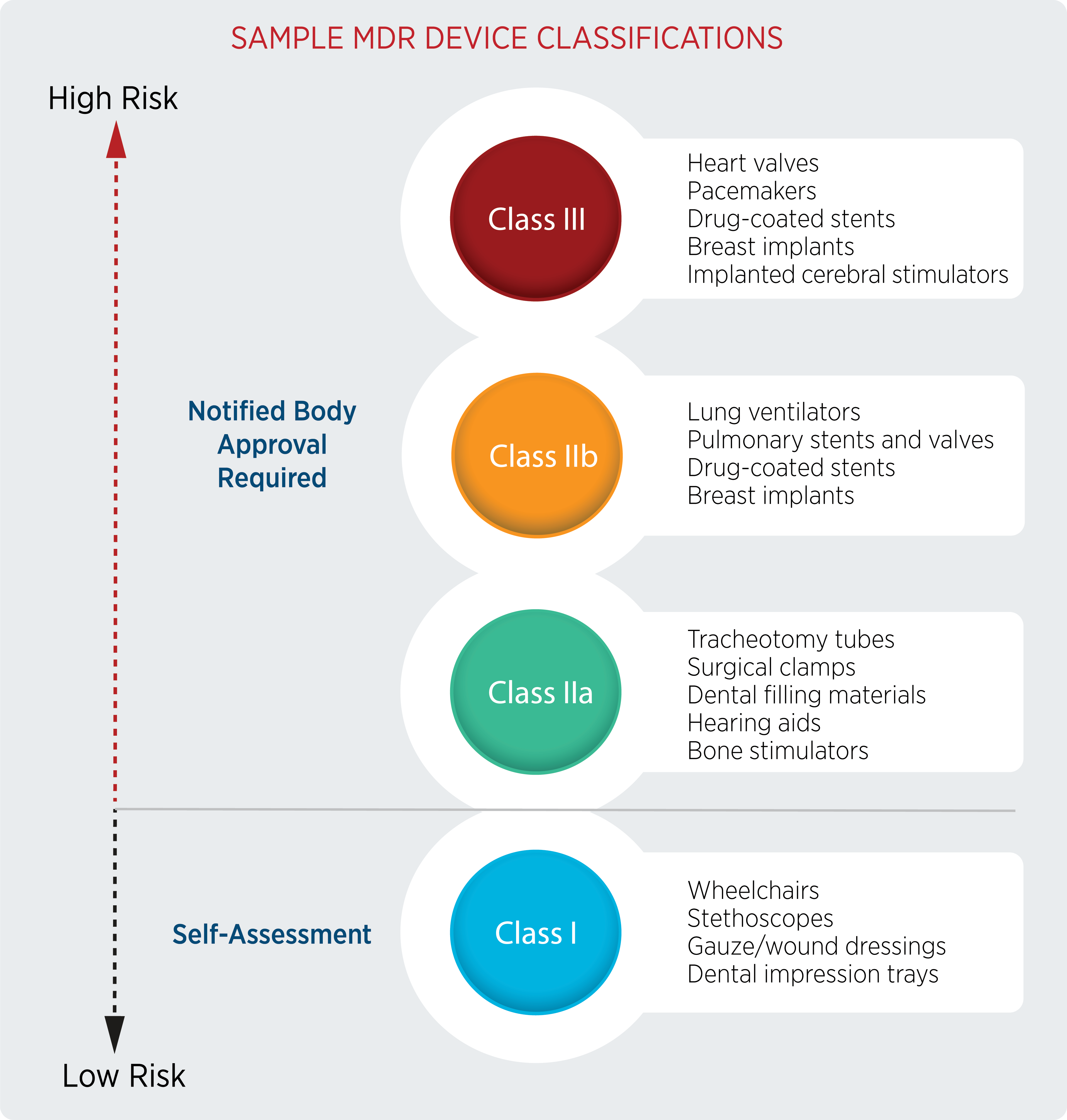
Prioritizing Devices for Certification
The next step is prioritizing devices for certification by identifying products that fall under exception criteria and conducting a product portfolio assessment.
Under Article V of EU MDR, device manufacturers can sell CE marked devices with a valid MDD certificate until May 2024, with the following conditions and caveat:
- The manufacturer must comply with post-market surveillance, vigilance, and registration requirements under the new MDR
- The MDD certificate must have been renewed by May 2020
- MDD Class I (self-declared) devices are not eligible
Next, you’ll want to perform a product portfolio assessment for the devices that remain on the list. For instance, if you have a legacy product that will cost $100,000 to bring into compliance but only generates $300,000 in annual revenue, you have a tough decision to make based on the product viability. You may have to pick and choose where to invest your resources.
While many larger companies conduct product reviews on an ongoing basis, it may be a more difficult exercise for smaller organizations.
IVDR Timeline Planning
Fortunately for companies that market IVDs, the new IVDR implementation deadline has been pushed back a year to May 26, 2022—the same go-live date for all six modules of EUDAMED.
The delay is a welcome reprieve for manufacturers, but given the even tighter constraints around NB resources until relatively recently. The good news is that, as of December 2020, a full 21 NBs have been certified to the new IVDR, up from just four in September 2020.
However, many companies are subject to IVDR requirements for the first time ever, creating a considerable volume of work for those NBs. IVDR, for example, now requires conformity assessment for roughly 90% of IVDs on the market, according to BSI, compared with just 10% under the previous IVDD.
In summary, IVD companies need to prepare now, as there’s little more than a year until the compliance deadline will be here.
Bridging the Gap with an Automated QMS
Many larger companies got started on MDR several years ago. However, small- to medium-sized companies (and even some larger companies) may be behind the curve due to a lack of internal regulatory expertise or initiative. To solve the problem, many businesses are realizing the value of a quality management system (QMS) automation to get more done in less time.
The large-scale transition to remote work resulting from the pandemic has only accelerated the trend. With only essential staff in the office, exchanging printed documents, meeting for a quick review, and waiting for live signatures are gone. An electronic repository with a workflow solution to manage and track quality processes becomes essential to sustain the required speed of changes while working remotely (or in-office).
Keeping documents on shared server drives leaves room for errors and inefficiency. An automated QMS, on the other hand, allows you to:
- Create a single secure digital repository for all document types
- Immediately access the most recent version of documents
- Pull up what was changed and by who
- Control permissions on who can edit documents
- Streamline regulatory submissions through the integration of quality data and reporting
Furthermore, data quality and data integrity are becoming increasingly critical, which is yet another area where the QMS provides a significant advantage. To be truly audit-ready—and to minimize scrutiny from regulatory authorities—it’s absolutely essential to have data and documentation secure and in order.
Many device manufacturers have their pipeline full and are just waiting for an NB to work with as they take their products through the regulatory process. Even without NBs available to review products, it’s critical that companies continue doing their due diligence in meeting MDR and IVDR requirements. Quality management software can help do it more effectively and efficiently, keeping companies audit-ready while simplifying the regulatory submissions process.

Resources:
- European Commission, Notified bodies for EU MDR
- TÜV SUD, IVDR FAQs
- BSI, In Vitro Diagnostics Medical Devices