
June 3, 2026
In the larger organizations I worked for as a quality leader, supplier auditing was almost always calendar-driven. Sometimes supplier audits happened once a year; in others, they might happen twice. I’d build the schedule to accommodate that, and the system would just run.
Over time, I shifted to a risk-based approach where how deeply and how often I audited a supplier wasn’t based on a calendar date, but rather on individual supplier risk.
How you determine high versus low risk is critical, and a key focus for regulators under FDA’s new Quality Management System Regulation (QMSR).
Let’s take a closer look at the benefits of risk-based supplier audits and the data that supports them, plus what FDA and registrars look for when evaluating your supplier program.
Learn how technology can support your supplier quality program with our free webinar, From Onboarding to Auditing: The Right Tools for Supplier Quality
What are some drawbacks of calendar-based supplier audits?
Standard calendar-based auditing is inefficient compared with risk-based audits, while also increasing the chances you’ll miss problems and run into issues with regulators.
The biggest drawbacks of this approach are:
- Wasted resources: When every supplier gets the same level of scrutiny, deep dives often land on low-risk suppliers that don’t actually need it.
- Missed signal on high-risk suppliers: When you audit high-risk suppliers at the same intensity as low-risk ones, time gets spread evenly across an unevenly risky population. This makes it more likely that something will fall through the cracks.
- False sense of security at the leadership level: If you visit suppliers annually, no one is watching high-risk suppliers in the 11 months between scheduled audits.
- Regulatory risk: Under FDA’s QMSR effective February 2026, the agency can now inspect supplier audit reports and expects risk-proportionate supplier evaluation. ISO 13485 clause 7.4 requires the same.
How should manufacturers evaluate supplier risk?
Evaluating supplier risk effectively requires looking at the problem through two lenses.
The first is criticality, and the second is performance.
Criticality is about the role the supplier’s materials and components play in your operations. Factors here include:
- Component importance: How essential is this part or material to your finished product?
- Regulatory exposure: Would a failure or change at this supplier trigger reporting obligations or affect your regulatory standing?
- Safety impact: What’s at stake from a product safety perspective if this supplier’s product fails?
- Supply-base concentration: Is this a single-source supplier or one of several?
Performance metrics to evaluate include:
- On-time delivery: Are shipments arriving when expected, and how consistent is the pattern?
- Responsiveness: How quickly does the supplier acknowledge and address issues, reply to your communications, and proactively handle recurring problems?
- Supplier Corrective Action Request (SCAR) frequency: Are issues recurring on the same supplier, on the same root causes?
- CAPA effectiveness: Do corrective actions prevent issues from coming back?
- Audit score trends: Which direction are audit scores moving over time?
Looking at both criticality and performance is key to determining the appropriate audit schedule and depth, both from an operational and regulatory standpoint.
A supplier with poor performance that provides a critical part is your highest risk and earns the deepest audit. A supplier with strong performance that provides a non-critical part may receive a lighter audit. For suppliers in the middle, the combination of criticality and performance determines where you spend your time.
Supplier audit depth: calibrating trust to evidence
One way to look at risk-based supplier audits is that the depth of the audit should reflect how much trust the supplier has earned.
For instance, a low-risk supplier with a clean track record has earned a certain amount of trust, which often translates into a lighter audit. They have an SOP. They say they follow it. The audit history backs them up. With trust, you’re more likely to take that at face value and spend less time verifying every claim.
A high-risk supplier, on the other hand, hasn’t earned that same trust yet. In some cases, repeat issues have spent down whatever trust they may have once had. With these high-risk suppliers, you might be spot-checking individual training records and verifying that what they tell you is actually happening on their floor. The audit footprint expands or contracts based on what the data says.
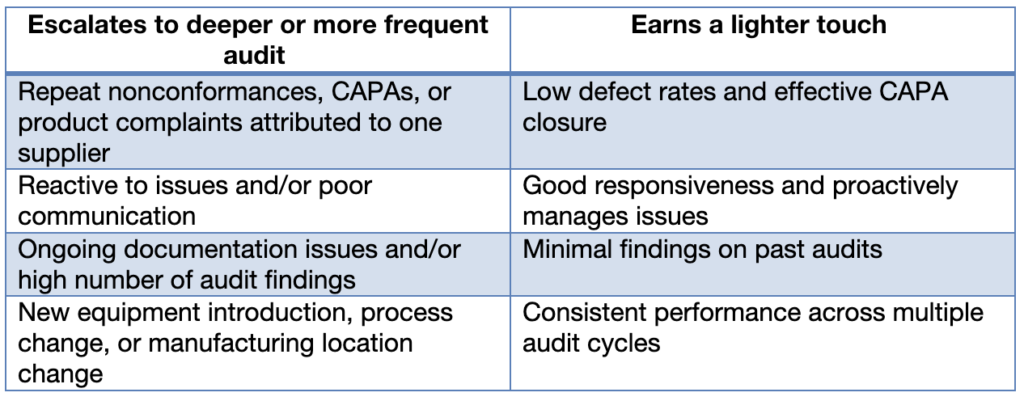
Supplier Audit Footprint
How data changes supplier audits
In my experience, once data drove the audit frequency, the work I did during the audit itself changed. I wasn’t there to confirm the SOP existed or check that training had been completed. I was there to figure out why three SCARs had clustered on the same training gap.
That kind of pattern is a signal that tells you where to dig deeper. In the training gap example, that could mean looking at things like:
- SOP clarity: If I were a production worker reading this SOP, would I understand what to do?
- Competency assessment: Is it a read-and-understand sign-off, or does it also have a quiz or exam the worker has to pass before training is marked complete?
- Root cause: Why are workers missing a step? Is it the SOP, training method, operating conditions, or something else?
Supplier performance data also shifts the tone of the audit as much as the scope. It’s harder for suppliers to get defensive when the data is in front of both of you. There’s no arguing over whether there’s actually an issue. Instead, the conversation moves to how you’ll solve it together.
What does the FDA want to see in your supplier audit documentation?
Under QMSR, FDA can now inspect supplier audit reports, and the previous QSR exemption for these records was removed. FDA expects manufacturers to maintain risk-based, ongoing control over suppliers under QMSR. ISO 13485:2016 also requires documented supplier evaluation and monitoring appropriate to the risks associated with the supplier and its products.
Across life sciences, regulators and ISO registrars are no longer taking “risk-based” at face value. They want to see the rationale behind it, which means they’re now looking at:
- Classification logic: How are you defining high-risk, medium-risk, and low-risk? What inputs go into that classification?
- Audit frequency logic: How does a supplier’s risk category translate to audit cadence?
- Escalation logic: What specific triggers move a supplier from routine monitoring to a deeper-dive audit?
- Underlying data: What performance metrics, CAPA records, complaint patterns, and trend analyses inform the classification?
- Trend analysis: Can you show that you are evaluating the trajectory of audit scores over multiple cycles?
The standard regulators hold manufacturers to is consistency between what your SOP says and what your data shows. If your SOP describes a risk-based program, the rationale, the metrics, and the audit decisions all need to line up with that description. Calling a program “risk-based” without the data to back it up isn’t enough.
How do you move to risk-based supplier audits?
Transitioning to risk-based supplier audits starts with program design and metrics, with an enterprise quality management system (EQMS) supporting the program once those decisions are made.
Best practices manufacturers should follow include:
- Defining your performance metrics: Decide what you’ll measure, and how: on-time delivery, responsiveness, SCAR frequency, CAPA effectiveness, audit score trends.
- Pulling the data together in one place: Patterns across CAPAs, nonconformances, audits, and other areas only become visible when systems share data. Paper-and-spreadsheet records can’t support the analysis you’ll need.
- Starting small: Pick one supplier group, like packaging suppliers, and test your approach. Look for patterns between nonconformances and audit performance. Calibrate as you go.
- Iterating on the metrics: As you collect data, correlations may reveal themselves. Low on-time delivery, for example, often correlates with product quality issues because suppliers skip steps to hit shipment dates.
- Reassessing classifications at least annually: Reassess suppliers as part of your management review. A supplier’s risk category should move as performance and exposure change.
Step 2 is where an EQMS becomes essential. A connected system linking CAPA, nonconformance, audit, complaint, and supplier quality data across modules gives you the visibility that a risk-based program is built on.
Three mistakes to avoid with risk-based supplier audits
There are several common mistakes quality teams should be aware of when making this shift:
- Calling your program risk-based without the data to back it up: Under QMSR and ISO 13485, this is now an inspection exposure. If you’re using the label, you need the documented logic and the supporting data underneath.
- Jumping into EQMS deployment before you understand the data you’re collecting: I see this often when ISO 13485 or 21 CFR Part 820 compliance is the trigger for implementing. Design the program first, get clear on what you’re measuring and why, then add the technology.
- Relying on a single metric: Audit score or on-time delivery alone doesn’t give you a balanced read. A supplier might score poorly on delivery, for example, and still run a strong nonconformance program.
The future of supplier audits
Expectations around supply chain quality are shifting, and supplier audit programs are shifting with them. Risk-based supplier auditing serves two purposes at once. It directs your time toward suppliers that actually need attention, while freeing up resources better spent improving processes than auditing suppliers with proven performance.
About the Author
Stephanie Ojeda is Vice President of Product Management for the Life Sciences industry at AssurX. Stephanie brings more than 18 years of leading quality assurance functions in a variety of industries, including pharmaceutical, biotech, medical device, food & beverage, and manufacturing.